From Undergrad Project to PLOS Cover: Using OpenACC for a Biophysics Problem
Something that started as a preliminary investigation for an undergraduate class project has become the cover of a prestigious journal publication. In the fall of 2017, two assistant professors from the University of Delaware—Sunita Chandrasekaran from the Department of Computer and Information Sciences and Juan Perilla from the Department of Chemistry and Biochemistry —were brainstorming research ideas related to molecular dynamics.
Perilla's lab focuses on the development of analysis methods driven by large-scale molecular dynamics simulations to highlight the utility of molecular dynamics simulations in relating atomic detail to the function of supramolecular complexes. This task cannot be achieved by smaller-scale simulations or existing experimental approaches alone. Perilla mentioned a code called PPM_One [2], which is used to predict chemical shifts of protein structures. These calculations provide atomic level information for each amino acid within a protein or protein complex and are valuable tools to help decipher the structure or secondary structures of molecules. However, due to the complexity of such calculations, the code was not viable for computing large complexes as it would take hours to run on a single protein complex.
It was a worthy problem to tackle and computer science and engineering undergraduate students, Eric Wright and Mauricio Ferrato, working in Chandrasekaran’s Computational and Research Programming Lab, were up to the challenge.

Approaching the Problem
As students in Chandrasekaran’s Vertically Integrated Projects (VIP)-HPC class, a unique undergraduate project-based course that focuses on high performance computing (HPC), Wright and Ferrato were tasked with investigating this PPM_One code base in order to determine if the code was suitable for accelerators and evaluate the effort needed to port this code to run on a heterogeneous HPC system that utilizes GPUs. Having limited experience in the realms of chemistry and HPC, graduate students were brought in to act as mentors for this project. Computational chemist Alex Bryer came to the table to help explain what the code is doing from a domain science perspective and how acceleration of the code would impact real-world experimentation [4]. At Professor Chandrasekaran’s request, I provided my GPU programming expertise as high-level GPU programming was one of the major focuses of my research in her group.
Having also mentored at several GPU hackathons organized by NVIDIA and Oak Ridge Leadership Computing Facility (OLCF) at Oak Ridge National Laboratory (ORNL), I was familiar with the process of profiling unfamiliar code, identifying hotspots that could be parallelized, and porting them to GPUs. We decided to use OpenACC [3] to port PPM_One to a heterogeneous system equipped with CPUs and GPUs because of its inherent portability. Our goal was to maintain a single code base for both CPUs and GPUs.
Our initial investigation into the code revealed some flaws that needed to be addressed before we could begin porting this code. We had to refactor this code in a number of ways in order to eliminate redundant/erroneous memory copies, inefficient list filtering, and the use of C++ STL containers like std::vector, which are not supported by OpenACC. Once these modifications were made to the original serial code, we were able to begin the cyclical process of profiling, identifying a computational bottleneck, accelerating that bottleneck, and beginning the process again to identify the next bottleneck.
The Results
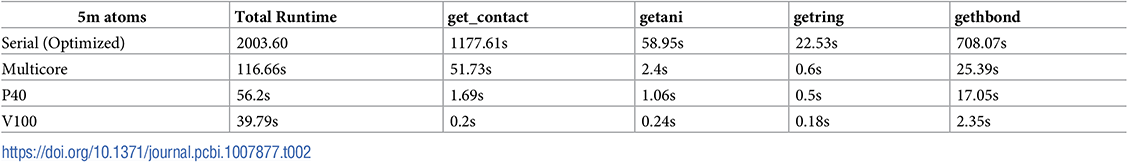
The results were impressive. Wright and Ferrato had taken a code that previously took 14 hours to run on a somewhat large protein complex (11.3 million atoms) using a single CPU core, ported it to the GPU using OpenACC, and ran it on a single NVIDIA V100 GPU for an accelerated runtime of just under 47 seconds.
The testing environment used to gather the results presented in this work was primarily NVIDIA’s PSG cluster, specifically PSG’s DGX-1b compute node, which consists of an Intel Xeon e5-2698 v4 20-core CPU paired with a Tesla V100 GPU. We also used a similar node that contained a P40 GPU instead of the V100 to obtain results on more than one GPU hardware architecture and demonstrate portability. The datasets we examined included the following sizes: 100,000, 1.5 million, 5 million, 6.8 million, and 11.3 million atoms. The best speedup was achieved on the largest dataset, allowing us to conclude that our code scales well as the problem size increases.
Final Thoughts
This project is a poster child of the VIP-HPC undergraduate course. Wright and Ferrato not only learned a great deal about parallel computing as undergraduate students, but this project played a major role in their decision to pursue research. Both are pursuing their PhDs in Chandrasekaran’s Computational Research and Programming Lab. Wright is currently working on a project with NCAR exploring acceleration of simulations of the Sun. Ferrato is working on a project with Nemours Alfred I. duPont Hospital for Children where he applies deep learning techniques to build predictive models to study pediatric oncology and its relapse. I have since graduated with my PhD and joined NVIDIA as a Solutions Architect primarily supporting the Department of Energy (DoE) and the Summit supercomputer at ORNL. Bryer is a PhD student studying the varying aspects of protein structures and the development of new methods and tools for biomolecular investigation under the supervision of Perilla.
Today, this work has been accepted for publication in the prestigious PLOS Computational Biology journal [1]. This work also won the 2019 ISC Best Research Poster award in the HPC category along with the 2018 VIP Mid Atlantic Best Research Poster award. Chandrasekaran and team have presented this work at GTC 2019, PASC 2019, and SIAM CSE 2019. The authors are immensely grateful to NVIDIA.
Read the full paper in the PLOS Computational Biology journal.
References:
1) Eric Wright, Mauricio Ferrato, Alexander J. Bryer, Robert Searles, Juan R. Perilla and Sunita Chandrasekaran. Accelerating prediction of chemical shift of protein structures on GPUs: Using OpenACC. PLOS Computational Biology. May 2020 DOI: https://doi.org/10.1371/journal.pcbi.1007877
2) Da-Wei Li and Rafael Brüschweiler. PPM: a side-chain and backbone chemical shift predictor for the assessment of protein conformational ensembles. Journal of Biomolecular NMR. Volume 54. pages 257–265. September 2012. DOI: https://doi.org/10.1007/s10858-012-9668-8
3) Sunita Chandrasekaran and Guido Juckeland. OpenACC for Programmers: Concepts and Strategies. Addison-Wesley Professional. ISBN-10: 0134694287. 2017.
4) Alexander J. Bryer, Jodi A. Hadden-Perilla, John E. Stone, and Juan R. Perilla. High-Performance Analysis of Biomolecular Containers to Measure Small-Molecule Transport, Transbilayer Lipid Diffusion, and Protein Cavities. Journal of Chemical Information and Modeling. V 59. no. 10. pages 4328–4338. September 2019. DOI: ttps://doi.org/10.1021/acs.jcim.9b00324
Author
